Что такое болезнь Ниманна-Пика?
Болезнь Нимана-Пика (PDD, дефицит кислой сфингомиелиназы) — редкое прогрессирующее генетическое заболевание, относящееся к группе лизосомных болезней накопления, возникающее в результате дефицита фермента кислой сфингомиелиназы, необходимого для расщепления (метаболизма) жирового вещества ( липидов), называемых сфингомиелином. Вследствие этого в различных тканях организма (печени, селезенке, головном мозге, костном мозге) накапливаются сфингомиелин и другие вещества.
Болезнь Ниманна-Пика очень изменчива, возраст начала, конкретные симптомы и тяжесть заболевания могут широко варьироваться от человека к человеку, иногда даже в пределах одной семьи.
Это расстройство лучше всего рассматривать как спектр заболеваний. На тяжелом конце спектра находится фатальное нейродегенеративное заболевание, которое проявляется в младенчестве (болезнь Ниманна-Пика А [BNPA]). На легкой стадии у пациентов практически отсутствуют неврологические симптомы, и часто пациенты доживают до зрелого возраста (болезнь Ниманна-Пика, тип B [BNPV]). Существуют также непрямые формы расстройства. BPN вызывается мутациями в гене SMPD1 и наследуется по аутосомно-рецессивному типу.
Классификация
Существует три расстройства, известные как болезнь Ниманна-Пика, типы A, B и C. Эти расстройства первоначально были сгруппированы вместе со схожими симптомами, но теперь известно, что это разные заболевания. BNP типа A и B вызваны мутациями в гене SMPD1, которые приводят к дефициту специфического фермента — кислой сфингомиелиназы. Болезнь Ниманна-Пика типа С вызывается мутациями в одном из двух разных генов и не содержит дефицитного фермента. По данным NORD, BNP типа C в настоящее время признается отдельным заболеванием от BNP типов A и B.
BNP традиционно делят на две подгруппы:
- нейропатический (тип А);
- ненейропатический (тип В).
Нейронопатия относится к заболеваниям, которые повреждают клетки головного мозга (нейроны). Тип А обычно вызывает тяжелое нейродегенеративное заболевание в младенчестве, тогда как тип В обычно не считается неврологическим расстройством. Однако, поскольку случаи попадают между этими двумя крайностями, такое широкое обозначение может сбивать людей с толку. Некоторые исследователи используют заболевание типа В для обозначения всех легких и промежуточных форм заболевания, которые могут иметь неврологические последствия.
Признаки и симптомы болезни Ниманна-Пика
Поскольку болезнь Ниманна-Пика является очень вариабельным заболеванием, следует помнить, что у пострадавших не будут проявляться все симптомы, описанные ниже, и каждый случай уникален. У некоторых детей в раннем возрасте развиваются тяжелые, опасные для жизни осложнения; в других легкое заболевание может быть диагностировано только во взрослом возрасте. Родители должны поговорить с врачом своего ребенка о конкретных симптомах и общем прогнозе.
Болезнь Ниманна-Пика, тип А

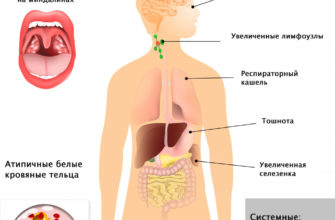
Тяжелую инфантильную форму ВЗК, известную как тип А, можно отличить от более легких форм, появляющихся в более позднем возрасте. Первичным симптомом в большинстве случаев у детей является аномальное увеличение печени и/или селезенки (гепатоспленомегалия), которое может прогрессировать до тех пор, пока печень и селезенка не станут объемными. Также может наблюдаться значительное скопление жидкости в брюшной полости (асцит). Пожелтение кожи и склер глаз (желтуха) может наблюдаться в младенчестве.
Дополнительные симптомы в младенчестве включают:
- проблемы с кормлением;
- запор;
- тошнота;
- рвота;
- желудочно-кишечный рефлюкс;
- раздражительность;
- потеря рефлексов;
- прогрессирующая потеря мышечного тонуса (гипотония).
Проблемы с питанием и другие аномалии (например, частая рвота) могут привести к отказу от здорового развития.
Накопление сфингомиелина в легких может привести к рецидивирующим респираторным инфекциям и затрудненному дыханию, что может привести к опасной для жизни легочной недостаточности.
У большинства детей развивается состояние, известное как пятна Пика (вишнево-красные пятна на глазах). Вишневые красные пятна влияют на пятно, область сетчатки, которая содержит чувствительные к свету клетки, необходимые для центрального зрения. Обычно он желтый. Пятницы PICA не всегда встречаются у людей, пострадавших от болезни.
Достижение основного этапа развития и общего развития может быть нормальным в первые несколько месяцев. Часто, однако, от 3 до 12 месяцев жизни развитие становится все сложнее, а пострадавшие дети теряют свои ранее приобретенные моторные навыки. У детей, страдающих заболеванием, глубокого неврологического ухудшения, увеличение мышечного тонуса и мышечная жесткость (спастичность) (спастичность), и может возникнуть болезнь фатальной.
Болезнь Ниманна-Пика, тип B

У людей с поздними формами ВЗК симптомы могут возникать как в младенчестве, так и в взрослой жизни. Иногда эти формы в совокупности называют болезнью бика не адгезионного типа Picka; Они обычно имеют более мягкий пробег, чем BNP -тип A (детская форма). Люди с мягкими формами могут жить до позднего взросления, а некоторые могут оставаться недиагностированными во взрослом возрасте. Болезнь бика типа Низнанны-B связана с системным заболеванием, которое может иметь очень различную тяжесть и частоту возникновения.
Hepatosplenicegalia является распространенным начальным симптомом и может покрываться от легких до тяжелых органов. Прогрессирующее увеличение селезенки может вызвать низкое количество тромбоцитов и лейкоцитов. Белые кровяные клетки (лейкоциты) помогают бороться с инфекциями, и их уменьшенное число может сделать человека, пораженного заболеванием, подверженным инфекциям. Кровавые тромбоциты — это специализированные клетки крови, которые позволяют организму образовывать сгустки и останавливать кровотечение. Уменьшенное количество тромбоцитов, называемых тромбоцитопении, может привести к эпизодам длительного кровотечения.
Увеличенная печень и селезенка могут вызвать боль в животе. Расширенной селезенке угрожают трещины, что может привести к жизненному опасному кровотечению в животе.
Большинство людей с болезнью типа В имеют определенную степень развития заболеваний печени. Большинство из них имеют аномальные шрамы в печени и печени. Шрамы могут возникать от легкого бессимптомного заболевания печени до явного цирроза и печеночной недостаточности.
Некоторые люди, которые пережили болезнь, наблюдают постепенное ухудшение функции легких. Для некоторых людей повреждение легких может быть легким, без заметных симптомов. У некоторых людей могут возникнуть затруднение дыхания во время упражнений (одышка). Другие люди могут иметь постоянное ухудшение дыхательной системы (легких) с серьезными ограничениями уровня активности и кислородной зависимости. Рецидивирующая пневмония может произойти.
У людей с покойным детьми IBD обычно нет неврологических симптомов, но легкие симптомы могут развиваться или в редких случаях значительные клинически неврологические симптомы. Некоторые люди, пострадавшие от детей и подростков, могут развиваться:
- Быстрые непроизвольные движения глаз (Nystagmus);
- Моторные расстройства, которые включают нестабильную походку и неуклюжость.
Интеллектуальная инвалидность и умственная отсталость тоже не возникают. Может быть:
- Волейбольные нарушения;
- НЕРВИГАТИЧЕСКИЕ НА НЕРНЫХ МЕМБРАНА, выстилающие заднюю часть глаза;
- периферическая невропатия.
Периферическая невропатия — это общий термин, определяющий все заболевания, влияющие на нервы за пределами центральной нервной системы. Общие симптомы включают потерю ощущения или дискомфорт, такие как зуд, сжигание или покалывание вдоль пораженных нервов.
Большинство детей, пострадавших от детей, ингибируются и недостаточный вес, хотя большинство из них, наконец, достигают почти нормального роста в взрослом. Может также возникнуть задержка сексуального созревания и созревания скелета. Остеопения (истончение костей) встречается у большинства людей.
Общие результаты у пострадавших включают аномальные уровни липидов в сыворотке (дислипидемия), особенно низкий уровень липопротеинов высокой плотности (холестерин ЛПВП, также известный как «хороший холестерин»), высокий уровень липопротеинов низкой плотности (холестерин ЛПНП, также известный как «хороший холестерин»). как «плохой холестерин») и высоким уровнем триглицеридов (гипертриглицеридемия).
Пострадавшие люди могут быть подвержены риску ранней ишемической болезни сердца.
Причины болезни Ниманна-Пика

Болезнь Нимана-Пика вызывается мутацией гена сфингомиелинфосфодиэстеразы-1 (SMPD1). Гены предоставляют инструкции для создания белков, играющих ключевую роль во многих функциях организма. Когда происходит генная мутация, белковый продукт может быть дефектным, неэффективным или отсутствовать. В зависимости от функции рассматриваемого белка это может повлиять на многие системы органов в организме, включая мозг.
Ген SMPD1 расположен на коротком плече (p) хромосомы 11 (11p15.4). Хромосомы, присутствующие в ядре клеток человека, несут генетическую информацию каждого человека. Клетки человеческого организма обычно имеют 46 хромосом. Пары хромосом человека пронумерованы от 1 до 22, а половые хромосомы помечены X и Y. У мужчин есть одна X и одна Y хромосома, а у женщин две X хромосомы. рука с надписью «p» «q».
Ген SMPD1 производит (кодирует) фермент, известный как кислая сфингомиелиназа. Мутация в этом гене вызывает недостаточный уровень функциональной копии фермента кислой сфингомиелиназы. Этот фермент необходим для расщепления (метаболизма) некоторых жировых веществ (липидов) в организме. Снижение или отсутствие активности фермента приводит к аномальному накоплению сфингомиелина в различных тканях организма. Сфингомиелин — это жировое вещество, которое содержится в большинстве клеточных мембран. Аномальное накопление сфингомиелина в определенных тканях организма вызывает признаки и симптомы болезни Нимана-Пика.
Болезнь Ниманна-Пика — это генетическое заболевание, которое передается по наследству от родителей и может поражать других членов семьи. Как правило, люди получают две копии большинства генов, одну от матери и одну от отца. Рецессивные генетические нарушения, такие как болезнь Нимана-Пика, возникают, когда человек наследует изменения или мутации в обеих копиях определенного гена (в данном случае гена SMPD1).
Если человек получит одну нормальную копию гена и одну измененную копию, он будет носителем болезни, но не будет проявлять ее признаков. Риск того, что два родителя-носителя передают дефектную копию гена и, следовательно, зачинают больного ребенка, составляет 25% при любой беременности. Риск рождения ребенка-носителя в качестве родителей составляет 50% для каждой беременности. Существует 25%-ная вероятность того, что ребенок получит нормальные копии гена от обоих родителей, не заболев и не носитель этой конкретной мутации. Риск одинаков для мужчин и женщин.
Затронутые группы населения
BNP одинаково влияет на мужчин и женщин. Точная заболеваемость и распространенность заболевания неизвестны, но оценивается в 1 случай на 250 000 человек в общей популяции. Однако, поскольку некоторые случаи неправильно диагностируются или вообще не диагностируются, трудно определить истинную распространенность среди населения в целом. Тяжелая инфантильная форма (тип А) может поражать разные этнические группы, но чаще встречается у лиц еврейского происхождения. Более поздние формы (тип Б) могут относиться ко всем этническим группам.
Схожие расстройства
Симптомы следующих нарушений могут быть похожи на симптомы BNP. Сравнение может быть полезным в дифференциальной диагностике.
Существует несколько типов метаболических нарушений, при которых происходит вторичное накопление определенных веществ, таких как жиры и углеводы, включая мукополисахаридоз (группа метаболических заболеваний соединительной ткани) и другие заболевания, связанные с лизосомальным накоплением. Эти расстройства включают:
- галактоземия;
- галактосиалидоз;
- сиалидоз;
- болезнь Ниманна-Пика типа С;
- болезнь Гоше;
- Болезнь Вольмана.
Диагностика
Диагноз заболевания основывается на констатации характерных симптомов, подробном анамнезе, тщательном клиническом обследовании и ряде специализированных обследований.
Лечение болезни Ниманна-Пика
Лечение болезни Ниманна-Пика может потребовать согласованных усилий группы специалистов. Педиатрам, неврологам, гепатологам, офтальмологам и другим специалистам в области здравоохранения может потребоваться систематическое и всестороннее планирование аффективного лечения ребенка. Также важна психологическая поддержка всей семьи. Генетическое консультирование может быть полезным для пострадавших и их семей.
Современные методы лечения нацелены на конкретные симптомы, которые испытывает каждый человек. При ВЗК типа А могут быть рекомендованы физическая и профессиональная терапия и периодическая оценка питания. Для обеспечения правильного питания может потребоваться имплантация питательного вещества (желудочного зонда). При этой процедуре через небольшой разрез в брюшной полости в желудок вводится тонкая трубка, что позволяет непосредственно принимать пищу и/или лекарство. Проблемы со сном, связанные с расстройством, можно лечить с помощью ночных седативных средств.
Некоторым взрослым с заболеванием типа В может потребоваться лечение дислипидемии (изменения уровня холестерина и/или триглицеридов). Нутритивная поддержка также рекомендуется людям с заболеванием типа B, чтобы обеспечить адекватное потребление калорий, достаточное потребление кальция и витамина D из-за риска остеопении, а также для снижения риска дислипидемии у взрослых.
Пациентам с болезнью Ниманна-Пика типа В, у которых наблюдается длительное кровотечение из-за тромбоцитопении, может потребоваться переливание крови. Людям с заболеваниями легких может потребоваться поддерживающая оксигенотерапия. Людям с увеличенной селезенкой рекомендуется избегать контактных видов спорта, чтобы избежать разрыва селезенки. У взрослых с гиперлипидемией следует контролировать уровень холестерина. Сообщалось также о трансплантации печени у некоторых взрослых с печеночной недостаточностью, вызванной BNP типа B.
Прогноз
Болезнь Ниманна-Пика типа А приводит к летальному исходу в раннем детстве. Клиническая картина и течение МНП типа А относительно однородны и характеризуются нормальным ребенком при рождении, сопровождающимся прогрессирующей гепатоспленомегалией с 3-месячного возраста и тяжелым нейродегенеративным течением, приводящим к смерти к 3-летнему возрасту.
В отличие от стереотипного фенотипа типа А, клиническая картина и течение у пациентов с болезнью BNP типа B более вариабельны в отношении клинических исходов, возраста начала заболевания и тяжести симптомов. Большинству пациентов диагноз ставится в младенчестве или детстве, когда при обычном физикальном обследовании обнаруживаются увеличенные печень и селезенка. Дети доживают до зрелого возраста с этим типом заболевания.
Ребенок с симптомами типа С до 1 года (хотя это другое заболевание) может не дожить до школьного возраста. Те, у кого есть симптомы, когда они идут в школу, могут дожить до подросткового возраста. Некоторые могут прожить до двадцати.