Что такое лизосомные болезни накопления?
Лизосомные накопительные заболевания (липоидоз) — это группа наследственных заболеваний обмена веществ, при которых вредные количества жировых веществ (липидов) накапливаются в различных клетках и тканях организма.
Люди с этими заболеваниями либо не вырабатывают достаточно одного из ферментов, необходимых для расщепления (метаболизма) липидов, либо вырабатывают ферменты, которые не работают должным образом.
Со временем это чрезмерное накопление жира может привести к необратимому повреждению клеток и тканей, особенно в головном мозге, периферической нервной системе (нервы от спинного мозга к остальному телу), печени, селезенке и костном мозге.
Что такое липиды?
Липиды представляют собой жироподобные вещества, которые являются важными элементами мембран внутри и между клетками, а также в миелиновой оболочке, которая покрывает и защищает нервы. Липиды включают масла, жирные кислоты, воски, стероиды (такие как холестерин и эстроген) и другие родственные соединения.
Эти жирные вещества естественным образом накапливаются в клетках, органах и тканях тела. Крошечные тела внутри клеток, называемые лизосомами, регулярно преобразуют или метаболизируют липиды и белки в более мелкие компоненты, чтобы обеспечить организм энергией. Заболевания, при которых внутриклеточный материал, который не может метаболизироваться, оставаясь в лизосомах, называют болезнями лизосомного накопления.
Помимо болезней накопления липидов, к другим болезням, связанным с накоплением липидов, относятся муколипидозы, при которых клетки и ткани накапливают чрезмерное количество липидов с прикрепленными к ним молекулами сахара, и мукополисахаридозы, при которых накапливаются чрезмерные количества больших сложных молекул сахара.
Как наследуются липоидозы?
Заболевания лизосомного накопления наследуются от одного или обоих родителей, которые несут дефектный ген, который регулирует определенный фермент, метаболизирующий липиды, в классе клеток организма. Их можно унаследовать двумя способами:
- Аутосомно-рецессивное наследование происходит, когда оба родителя являются носителями и передают копию дефектного гена, но ни один из родителей не страдает заболеванием. Каждый ребенок, рожденный от этих родителей, имеет 25% шанс унаследовать обе копии дефектного гена, 50% шанс быть носителем, как и их родители, и 25% шанс не унаследовать какую-либо копию дефектного гена. Дети обоих полов могут быть предрасположены к аутосомно-рецессивному типу наследования.
- Х-сцепленное (или сцепленное с полом) рецессивное наследование происходит, когда мать несет пораженный ген на Х-хромосоме. Х- и Y-хромосомы участвуют в определении пола. У женщин две хромосомы X, а у мужчин одна хромосома X и одна хромосома Y. Сыновья женщин-носителей имеют 50-процентную вероятность унаследовать и затронуть это заболевание, потому что сыновья получают один X от своей матери и один Y от отца. Дочери имеют 50-процентный шанс унаследовать пораженную Х-хромосому от своей матери и являются носителями или находятся в легкой форме. Больные мужчины не передают болезнь своим сыновьям, но их дочери будут носителями и будут передавать болезнь.
Типы и симптомы лизосомных болезней накопления
Болезнь Гоше — самое распространенное из лизосомальных заболеваний. Заболевание вызвано дефицитом фермента глюкоцереброзидазы, что приводит к накоплению глюкоцереброзида во многих тканях. Жировое вещество может накапливаться в головном мозге, селезенке, печени, почках, легких и костном мозге.

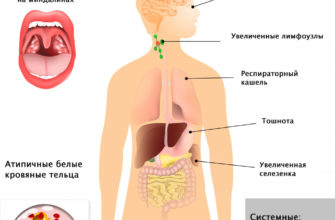
Симптомы могут включать повреждение головного мозга, увеличение селезенки и печени, нарушение функции печени, костные заболевания и повреждение костей, которые могут вызывать боль и переломы, увеличение лимфатических узлов и (иногда) соседних суставов, газы, коричневатую кожу, анемию, низкое количество тромбоцитов. и желтые пятна в глазах.
Те, кто пострадали наиболее серьезно, также могут быть более подвержены инфекциям. Это заболевание одинаково поражает мужчин и женщин.
Существует три общих клинических подтипа болезни Гоше:
- Тип 1 (или ненейронопатический тип) — наиболее распространенная форма заболевания в США и Европе. Мозг не поражается болезнью, но могут возникать изменения в легких и, реже, в почках. Симптомы могут проявиться в молодом или зрелом возрасте и включать увеличение печени и сильно увеличенную селезенку, что может привести к разрыву и дополнительным осложнениям. Слабость скелета и болезни костей могут быть обширными. У людей этой группы обычно легко появляются синяки из-за низкого количества тромбоцитов в крови. Они также могут чувствовать усталость от анемии. В зависимости от начала и тяжести заболевания люди с типом 1 могут дожить до взрослого возраста. У многих людей симптомы легкие или могут отсутствовать совсем. Хотя болезнь Гоше 1 типа распространена среди людей еврейского происхождения ашкенази, она может поражать людей любой национальности.
- Тип 2 (или острая педиатрическая болезнь Гоше) обычно начинается в течение 3 месяцев после рождения. Симптомы включают обширное и прогрессирующее повреждение головного мозга, спастичность, судороги, жесткость конечностей, увеличение печени (гепатомегалия) и селезенки (спленомегалия), аномальные движения глаз, а также плохую способность сосать и глотать. Больные дети обычно умирают в возрасте до 2 лет.
- Тип 3 (хроническая нейронопатическая форма) может начаться в любое время в детстве и даже во взрослом возрасте. Он характеризуется медленно прогрессирующими, но менее тяжелыми неврологическими симптомами по сравнению с острой болезнью Гоше 2 типа. Основные симптомы включают нарушения движений глаз, когнитивный дефицит, плохую координацию, судороги, увеличение селезенки и / или печени, а также нарушения скелета, заболевания крови. в том числе анемия и проблемы с дыханием. Почти каждый человек с болезнью Гоше 3 типа, получающий заместительную ферментную терапию, достигнет совершеннолетия.
У пациентов с типами 1 и 3 заместительная ферментная терапия, вводимая внутривенно каждые две недели, может значительно уменьшить размер печени и селезенки, уменьшить скелетные аномалии и обратить другие симптомы. Успешная трансплантация костного мозга устраняет неврологические симптомы заболевания. Однако эта процедура связана с высоким риском и редко выполняется у людей с болезнью Гоше.
В редких случаях может потребоваться операция для полного или частичного удаления селезенки (если у человека очень низкое количество тромбоцитов или увеличенный орган значительно влияет на самочувствие человека). Некоторым пациентам с анемией может быть полезно переливание крови. Другим может потребоваться операция по замене сустава, чтобы улучшить подвижность и качество жизни. В настоящее время не существует эффективного лечения повреждения мозга, которое может возникнуть у людей с болезнью Гоше 2 и 3 типа.
Болезнь Ниманна-Пика — это группа аутосомно-рецессивных заболеваний, вызванных накоплением жира и холестерина в клетках печени, селезенки, костного мозга, легких и, в некоторых случаях, головного мозга.

Неврологические осложнения могут включать атаксию (отсутствие мышечной координации, которая может влиять, среди прочего, на ходьбу, письмо и прием пищи), паралич глаз, дегенерацию мозга, трудности в обучении, спастичность, трудности с кормлением и глотанием, невнятную речь, потерю мышечного тонуса, гиперчувствительность к мышцам. прикосновение и некоторые помутнения роговицы из-за скопления избыточного материала. У 50% людей, страдающих этим заболеванием, вокруг центра сетчатки появляется характерный вишнево-красный ореол, который врач может увидеть с помощью специального инструмента.
Болезнь Ниманна-Пика делится на три основных подтипа:
- Тип А, наиболее тяжелая форма, начинается в раннем детстве. Младенцы выглядят нормальными при рождении, но к 6 месяцам у них развиваются глубокие повреждения головного мозга, увеличиваются печень и селезенка, а также увеличиваются лимфатические узлы и шишки под кожей (ксантомы). Селезенка может увеличиться в 10 раз по сравнению с нормальным размером и может лопнуть, что приведет к кровотечению. Эти дети слабеют, теряют двигательные функции, могут страдать анемией и склонны к рецидивам инфекций. Они редко живут дольше 18 месяцев. Эта форма болезни чаще встречается в еврейских семьях.
- Тип B (или ювенильное начало) обычно не влияет на мозг, но у большинства детей развивается атаксия, повреждение нервов, выходящих из спинного мозга (периферическая невропатия), и проблемы с легкими, которые прогрессируют с возрастом. Увеличение печени и селезенки часто встречается у подростков. Люди с типом B могут прожить относительно долгую жизнь, но многим требуется пожизненная кислородная терапия при повреждении легких. Типы Ниманна-Пика A и B возникают в результате накопления жирного вещества, называемого сфингомиелином, из-за дефицита фермента, называемого сфингомиелиназа.
- Тип C может возникнуть в молодом возрасте или развиться в подростковом и даже во взрослом возрасте. Болезнь Ниманна-Пикатипа С вызывается не дефицитом сфингомиелиназы, а дефицитом белков NPC1 или NPC2. В результате различные липиды, особенно холестерин, накапливаются в нервных клетках и вызывают сбои в их работе. Поражение мозга может быть обширным, приводя к неспособности смотреть вверх и вниз, затруднениям при ходьбе и глотании, прогрессирующей потере слуха и прогрессирующей деменции. У людей с типом С наблюдается лишь незначительное увеличение селезенки и печени. Продолжительность жизни у людей с типом С сильно различается. Некоторые люди умирают в детстве, в то время как другие, которые кажутся менее пораженными, могут дожить до взрослой жизни.
В настоящее время нет лекарства от болезни Ниманна-Пика. Лечение поддерживающее. Дети обычно умирают от инфекции или прогрессирующего неврологического упадка. Сообщалось о трансплантации костного мозга у нескольких пациентов с типом B с неоднозначными результатами.
Болезнь Фабри, также известная как дефицит альфа-галактозидазы A, вызывает накопление жирных веществ в вегетативной нервной системе (часть нервной системы, которая контролирует непроизвольные функции, такие как дыхание и сердцебиение), глазах, почках и сердечно-сосудистой системе.

Болезнь Фабри — единственное заболевание липидов, сцепленное с Х. Заболевание в основном поражает мужчин, хотя более легкая и вариабельная форма встречается у женщин. Иногда у пораженных женщин наблюдаются тяжелые симптомы, подобные тем, которые наблюдаются у мужчин с этим заболеванием. Симптомы обычно появляются в детском или подростковом возрасте.
Неврологические симптомы включают жгучую боль в руках и ногах, которая усиливается в жаркую погоду или после упражнений, а также скопление избыточного материала в прозрачных слоях роговицы (что приводит к помутнению, но без изменений зрения). Жир, который накапливается в стенках кровеносных сосудов, может нарушить кровообращение, подвергая человека риску инсульта или сердечного приступа. Другие симптомы включают увеличение сердца, прогрессирующую почечную недостаточность, приводящую к терминальной почечной недостаточности, желудочно-кишечные проблемы, снижение потоотделения и лихорадку.
Ангиокератомы (маленькие доброкачественные красно-пурпурные пятна на коже) могут развиваться в нижней части туловища и с возрастом становятся более многочисленными.
Люди с болезнью Фабри часто умирают преждевременно от осложнений, вызванных сердечной недостаточностью, почечной недостаточностью или инсультом. Такие лекарства, как фенитоин и карбамазепин, часто назначают для снятия боли при болезни Фабри, но не для ее лечения. Метоклопрамид или липисорб (пищевая добавка) могут облегчить желудочно-кишечные проблемы, которые часто встречаются у людей с болезнью Фабри, а некоторым людям может потребоваться пересадка почки или диализ. Замена ферментов может уменьшить накопление, облегчить боль и сохранить функцию органов у некоторых людей с болезнью Фабри.
Болезнь Фарбера, также известная как липогранулематоз Фарбера, описывает группу редких аутосомно-рецессивных заболеваний, вызывающих накопление жировых веществ в суставах, тканях и центральной нервной системе. От него страдают как мужчины, так и женщины. Заболевание обычно начинается в раннем детстве, но может возникнуть и в более позднем возрасте.
У детей с классической болезнью Фарбера в первые несколько недель жизни развиваются неврологические симптомы, которые могут включать повышенную сонливость и апатию, а также затруднение глотания. Также может произойти поражение печени, сердца и почек. Другие симптомы могут включать контрактуры суставов (хроническое укорочение мышц или сухожилий вокруг суставов), рвоту, артрит, увеличение лимфатических узлов, артрит, охриплость голоса и узлы под кожей, которые утолщаются вокруг суставов по мере прогрессирования заболевания.
Людям с проблемами дыхания может потребоваться установка дыхательной трубки. Большинство детей с этим заболеванием умирают в возрасте до 2 лет, обычно от болезни легких. При одной из самых тяжелых форм заболевания увеличение печени и селезенки может быть диагностировано вскоре после рождения. Младенцы, рожденные с этой формой заболевания, обычно умирают в течение 6 месяцев.
Болезнь Фарбера вызвана дефицитом фермента под названием церамидаза. В настоящее время не существует специального лечения болезни Фарбера. Для облегчения боли могут быть назначены кортикостероидные препараты. Пересадка костного мозга может уменьшить гранулемы (небольшие образования воспалительной ткани) у людей с легким воспалением или у пациентов без осложнений со стороны легочной или нервной системы. У пожилых людей гранулемы могут быть уменьшены или удалены хирургическим путем.
Ганглиозидоз состоит из двух разных групп генетических заболеваний. Оба заболевания передаются по аутосомно-рецессивному типу и одинаково влияют на мужчин и женщин.
GM1-ганглиозидоз.
GM1-ганглиозидоз вызывается дефицитом фермента бета-галактозидазы, что приводит к аномальному накоплению кислых липидных веществ, особенно в нервных клетках центральной и периферической нервной системы. GM1-ганглиозидоз характеризуется тремя клиническими признаками:
- GM1 (наиболее тяжелый подтип, возникающий вскоре после рождения) может включать нейродегенерацию, судороги, увеличение печени и селезенки, грубые черты лица, костные аномалии, жесткость суставов, вздутие живота, мышечную слабость, чрезмерную реакцию на раздражители и затруднения при ходьбе. Примерно у половины пострадавших появляются вишнево-красные пятна на глазах. Дети могут быть глухими и слепыми до 1 года и часто умирают до 3 лет от сердечных осложнений или пневмонии.
- GM1-поздний детский ганглиозидоз обычно начинается в возрасте от 1 до 3 лет. Неврологические симптомы включают атаксию, судороги, слабоумие и затрудненную речь.
- GM1-ганглиозидоз развивается в возрасте от 3 до 30 лет. Симптомы включают снижение мышечной массы (мышечное истощение), неврологические осложнения, которые менее серьезны и прогрессируют медленнее, чем при других формах заболевания, помутнение роговицы у некоторых людей и постоянные сокращения мышц, которые вызывают скручивание и повторяющиеся движения или нарушения осанки (дистония). . Ангиокератомы могут развиваться в нижней части туловища. Размер печени и селезенки у большинства больных нормальный.
GM2-ганглиозидоз.
GM2-ганглиозидоз также заставляет организм накапливать чрезмерное количество жирных кислот в тканях и клетках, в первую очередь нервных. Эти нарушения возникают в результате дефицита фермента бета-гексозаминидазы. Расстройства GM2 включают:
- Болезнь Тея-Сакса (также известная как вариант В-ганглиозидоза GM2) и ее вариантные формы вызываются дефицитом фермента гексозаминидазы А. Больные дети нормально развиваются в течение первых нескольких месяцев жизни. Симптомы начинаются в возрасте 6 месяцев и включают прогрессирующую потерю умственных способностей, деменцию, снижение зрительного контакта, усиление реакции на шум, прогрессирующую потерю слуха, ведущую к глухоте, затрудненное глотание, слепоту, вишнево-красные пятна на сетчатке и некоторый паралич. Судороги могут начаться на втором году жизни ребенка. Младенцы со временем могут стать зависимыми от зонда для кормления и часто умирают в возрасте до 4 лет от рецидивирующих инфекций. К сожалению, специального лечения нет. Первоначально противосудорожные препараты могут контролировать припадки. Другие поддерживающие методы лечения включают правильное питание и гидратацию, а также методы поддержания дыхательных путей на открытом воздухе. Редкая форма этого расстройства, называемая поздней формой болезни Тея-Сакса, возникает у людей в возрасте от 20 до 30 лет и характеризуется нестабильной походкой и прогрессирующим неврологическим ухудшением.
- Болезнь Сандхоффа (вариант AB) — тяжелая форма болезни Тея-Сакса. Начало обычно происходит после 6 месяцев жизни и не ограничивается какой-либо этнической группой. Неврологические симптомы могут включать прогрессирующее ухудшение работы центральной нервной системы, двигательную слабость, раннюю слепоту, выраженную звуковую реакцию, спастичность, мышечный шок или судороги (миоклонус), эпилепсию, аномально увеличенную голову (макроцефалия) и красные пятна в глазах. Другие симптомы могут включать частые респираторные инфекции, шумы в сердце, кукольные черты лица, а также увеличение печени и селезенки. Специфического лечения болезни Сандхоффа не существует. Как и в случае с болезнью Тея-Сакса, поддерживающая терапия включает постоянное поддержание дыхательных путей открытыми, а также правильное питание и гидратацию. Противосудорожные препараты могут первоначально контролировать эпилепсию (припадки).
Болезнь Краббе (также известная как глобоклеточная лейкодистрофия и галактозилцерамидный липидоз) — аутосомно-рецессивное заболевание, вызванное дефицитом фермента галактоцереброзидазы. Заболевание чаще всего поражает младенцев в возрасте до 6 месяцев, но может возникнуть в подростковом или взрослом возрасте.

Накопление непереваренного жира увеличивает защитную изолирующую оболочку нерва (миелиновую оболочку) и вызывает серьезные нарушения умственных и двигательных навыков. Другие симптомы включают мышечную слабость, снижение способности мышц к растяжению (гипотония), мышечное напряжение (спастичность), внезапное дрожание или подергивание конечностей (миоклонические припадки), раздражительность, необъяснимую лихорадку, глухоту, слепоту, паралич и затруднение глотания. Также может произойти длительная потеря веса.
Заболевание можно диагностировать с помощью ферментативных тестов и характерного скопления клеток в глобоидные тела в белом веществе мозга, демиелинизации и дегенерации нервов, а также разрушения клеток мозга. У младенцев болезнь обычно заканчивается летальным исходом к 2 годам. Люди с более поздней формой заболевания имеют более легкое течение болезни и живут намного дольше.
Специфического лечения болезни Краббе не разработано, хотя некоторым людям может помочь ранняя трансплантация костного мозга. Люди с более поздней формой заболевания имеют более легкое течение болезни и живут намного дольше.
Метахроматическая лейкодистрофия, или МЛД, представляет собой группу заболеваний, характеризующихся накоплением жировых веществ в белом веществе центральной нервной системы и периферических нервов и, в некоторой степени, в почках. Как и болезнь Краббе, MLD влияет на миелин, который покрывает и защищает нервы. Это аутосомно-рецессивное заболевание, вызванное дефицитом фермента арилсульфатазы А. Это заболевание поражает как мужчин, так и женщин.
Метахроматическая лейкодистрофия имеет три отличительных формы: поздний младенец, подросток и взрослая.
- МЛД у позднего ребенка обычно начинается между 12 и 20 месяцами после рождения. Поначалу дети могут казаться нормальными, но у них возникают трудности с ходьбой и тенденция к падению, сопровождающаяся перемежающейся болью в руках и ногах, прогрессирующей потерей зрения, ведущей к слепоте, задержкам развития и потере ранее достигнутых вех, дисфагии, судорогам и слабоумию вплоть до 2 года. Дети также постепенно теряют мышцы, ослабевают и в конечном итоге теряют способность ходить. Большинство детей с этой формой заболевания умирают в возрасте до 5 лет.
- Юная MLD обычно начинается в возрасте от 3 до 10 лет. Симптомы включают снижение успеваемости в школе, снижение умственной работоспособности, атаксию, судороги и слабоумие. Симптомы прогрессируют, смерть наступает через 10-20 лет после начала заболевания.
- Взрослая форма. Симптомы у взрослых начинаются после 16 лет и могут включать атаксию, судороги, аномальный тремор, трудности с концентрацией внимания, депрессию, психические расстройства и слабоумие. Смерть обычно наступает через 6-14 лет после появления симптомов.
MLD нельзя вылечить. Лечение симптоматическое и поддерживающее. В некоторых случаях трансплантация костного мозга может отсрочить прогрессирование заболевания. Значительные успехи были достигнуты в генной терапии на животных моделях MLD и в клинических испытаниях.
Болезнь Вулмана, также известная как дефицит лизосомальной кислотной липазы, представляет собой тяжелое нарушение накопления липидов, которое обычно заканчивается смертельным исходом в возрасте 1 года. Это аутосомно-рецессивное заболевание, характеризующееся накоплением сложных эфиров холестерина (обычно транспортной формы холестерина) и триглицеридов (химическая форма, в которой жир находится в организме), которые могут значительно накапливаться и повредить клетки и ткани.
Этим заболеванием страдают как мужчины, так и женщины. Младенцы нормальные и активные при рождении, но быстро развивается постепенное ухудшение умственного развития, увеличивается печень и сильно увеличивается селезенка, вздутие живота, желудочно-кишечные проблемы, желтуха, анемия, рвота и отложения кальция в надпочечниках, вызывающие их затвердевание.
Другой тип недостаточности лизосомальной кислой липазы — это болезнь накопления сложных эфиров холестерина. Это чрезвычайно редкое заболевание возникает в результате накопления сложных эфиров холестерина и триглицеридов в клетках крови, лимфы и лимфоидной ткани. У детей печень увеличивается в зрелом возрасте, что приводит к циррозу и хронической печеночной недостаточности. У детей также могут образовываться отложения кальция в надпочечниках и развиваться желтуха на более поздних стадиях заболевания.
Замещение ферментов активно изучается как при болезни Вольмана, так и при накоплении эфиров холестерина.