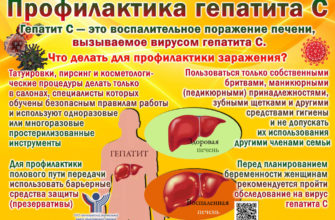
Что такое метахроматическая лейкодистрофия?
Метахроматическая лейкодистрофия (MLD) — это унаследованное заболевание, характеризуемое накоплением клеток, называемых жирами сульфатыдами. Это особенно влияет на накопление клеток нервной системы, которые производят миелину, изоляционный материал, который защищает нервы. Нервные клетки с покрытием миелина образуют ткань, известную как белый вещество. Накопление сульфид-продуцирующих клеток миелина вызывает прогрессирующее разрушение белого вещества (лейкодистрофию) по всей нервной системе, включая мозг и спинного мозга (центральная нервная система), а нервы, соединяющие мозг и спинного мозга, мышцы и клетки чувств, которые обнаружить такие впечатления , как прикосновение, боль, тепло и звук (периферическая нервная система).
У людей с млагородным повреждением белой материи приводит к постепенному ухудшению интеллектуальной функции и двигательных навыков, таких как способность ходить. Жертвы также возникают потери ощущения в конечностях (периферическая недержание), недержание мочи, эпилепсия, паралич, неспособность говорить, слепоту и потерю слуха. В итоге пациенты теряют осведомленность об окружающей среде и прекращают отвечать. Хотя неврологические проблемы являются основной особенностью метахроматической лейкодистрофии, описывают влияние накопления сульфида на другие органы и ткани, чаще всего на желчном пузыре.
Это расстройство известно как метакроматическое лейкодистрофию, поскольку при взгляде под микроскопом накопление сульфида в клетках проявляется как гранулы, которые не окрашены так же, как и другие клеточные материалы (метакроматические). Лейкодистрофия — это генетическое расстройство, которое ухудшает миелинина в мозге.
Признаки и симптомы
Каждый из подтипов MLD имеет специфические симптомы и скорость прогрессирования. Каждый из подтипов на основе возраста начала заболевания.
Наиболее распространенной формой метахроматической лейкодистрофии, происходящей примерно в 50-60% всех людей с этим расстройством, называется формой PóźnoNiemowlęcą (Inflile). Эта форма расстройства обычно встречается во втором году жизни. Пострадавшие дети проигрывают развитую речь, становятся слабыми и испытывают трудности прогулки (походное беспокойство). С расстройствами прогрессирования мышечное напряжение обычно уменьшается первым, а затем увеличивается до жесткости. Люди с поздней, инфантильной формой метахроматической лейкодистрофии обычно не выживают детства.
Через 20-30 процентов людей с метахроматическим лейкодистрофией происходит в возрасте от 4 лет и половое созревание. В этом воплощении несовершеннолетних первые признаки беспокойства могут быть поведенческие проблемы и растущие трудности в данной области. Расстройство прогрессирования медленнее, чем в конце детства, а затронутые могут жить до 20 лет после диагностики.
Взрослая форма метахроматической лейкодистрофии затрагивает от 15 до 20 процентов людей с этим расстройством. В этой форме первые симптомы появляются во время подросткового возраста или позже. Часто первыми симптомами являются поведенческие проблемы, такие как алкоголизм, злоупотребление веществом или трудности в школе или работе. В пострадавшем человеке могут испытывать психические симптомы, такие как заблуждения или галлюцинации. Люди со взрослой формой расстройства могут жить через 20-30 лет после диагностики. В это время могут быть периоды относительной стабильности и в другие времена быстрее снижение.
Причины
УСА является генетическим заболеванием аутосомно-рецессивной. Рецессивные генетические расстройства возникают, если обе копии гена затронуты. Если ребенок зависит, в большинстве случаев, родители являются носителями, что означает, что каждый родитель будет иметь модифицированный (мутантный) и один нормальную копию гена ASA, но не будет иметь никаких симптомов. Риск передачи дисфункциональных носителей генов обоих родителей, и, таким образом, рождение больного ребенка, в каждой беременности 25%. Риск того, чтобы иметь ребенка, который является носителем, например, родителей, составляет 50% при каждой беременности. Вероятность того, что ребенок будет получать правильные гены от обоих родителей 25%. Риск одинаков для мужчин и женщин. Ген ASA кодирует протеин арилсульфатазу А (ARSA). Существует связь между определенными мутациями и подтипом MLD (корреляция генотип-фенотип). В редких случаях дети с MLD имеют функциональные копии гена ASA, но дефект гена PSAP, который кодирует несколько белков сапозина, включая сапозин B, активатор ASK.
Аномалии в этих белках приводят к неспособности организма расщеплять жиры (липиды), содержащиеся в сульфатах (сульфатидах). Сульфид накапливается в нервной системе, почках, яичках и головном мозге, препятствуя выработке миелина, вещества, которое изолирует и защищает нервы. Когда сульфат накапливается в нервной системе, миелин разрушается, и нервы, соединяющие головной и спинной мозг, не функционируют должным образом. Это приводит к проблемам с функционированием головного мозга, что, в свою очередь, приводит к психическим и физическим проблемам у людей с МЛД. Симптомы варьируются в зависимости от того, какие части мозга поражены.
Затронутые группы населения
Истинная распространенность метахроматической лейкодистрофии неизвестна, но, по оценкам, она составляет от 1 на 40 000 до 1 на 160 000. Уровень заболеваемости навахо выше — 1 на 2500 человек. В некоторых популяциях на Ближнем Востоке эти цифры могут быть еще выше. Гендерных различий не обнаружено.
Близкие по симптомам расстройства
Существует множество родственных заболеваний, которые имеют схожие причины или симптомы, включая другие лейкодистрофии, которые могут иметь сходные проявления в раннем детстве.
- Псевдоселекция алисульфатазы А включает низкую активность фермента ARSA (менее 15% от нормы), но не имеет симптомов.
- Повторный дефицит сульфатазы вызывается потерей (частичной или полной) всех сульфатаз, включая ARSA. Дети с метахроматической лейкодистрофией будут иметь низкую ферментативную активность многих сульфатаз, а не только ARSA.
- Приобретенные заболевания, такие как воспалительная демиелинизирующая полинейропатия, могут выглядеть сходными при первоначальном электрофизиологическом исследовании. МЛД следует учитывать при дифференциальной диагностике у детей раннего возраста с диагнозом хроническая воспалительная демиелинизирующая полинейропатия или синдром Гийена-Барре (СГБ). Эти диагнозы могут быть легко поставлены с помощью теста на сульфатазу.
Диагностика
Первое подозрение на МЛД возникает после выявления характерной тенденции к прогрессирующему ухудшению самочувствия. При поздней инфантильной форме затруднение при ходьбе часто является первым симптомом, который может проявляться новой неспособностью полностью поднять ноги при ходьбе. В случае MLD у взрослых первыми признаками являются невнятная речь и поведенческие проблемы, которые включают трудности в школе, изменения поведения и снижение способности к обучению. У подростков с MLD могут быть двигательные или когнитивные симптомы.
— Испытания и клинические испытания.
Диагноз MLD ставится на основании генетических и биохимических тестов. Генетическое тестирование может выявить мутации в генах ASA и PSAP. Биохимические исследования включают активность фермента сульфатазы и экскрецию сульфатидов с мочой.
Магнитно-резонансная томография (МРТ) может подтвердить диагноз MLD. МРТ показывает изображение человеческого мозга и может показать наличие или отсутствие миелина. Существует классический образец потери миелина в мозге людей с MLD. По мере прогрессирования заболевания визуализирующие исследования показывают накопление повреждений головного мозга. Примечательно, что у маленьких детей первоначальные результаты тестов визуализации мозга могут быть нормальными.
Стандартные методы лечения
В настоящее время не существует лечения или лечения метахроматической лейкодистрофии у симптоматических пациентов поздней инфантильной формы или у подростков и взрослых с тяжелыми симптомами. Эти пациенты обычно получают клиническое лечение боли и симптомов.
У бессимптомных пациентов с МЛД в позднем детстве, а также у пациентов с ювенильной или взрослой метахроматической лейкодистрофией, у которых наблюдаются бессимптомные или легкие симптомы, может быть рассмотрена трансплантация костного мозга (включая трансплантацию стволовых клеток), что может замедлить прогрессирование заболевания в центральная нервная система нервная система. Однако результаты для периферической нервной системы были менее драматичными, а долгосрочные результаты этих видов лечения неубедительны. Совсем недавно было показано, что удаление стволовых клеток из костного мозга больных детей и заражение ретровирусом, замена мутировавшего гена стволовой клетки восстановленным геном перед повторным введением его пациенту, где они размножились. Все дети до пяти лет были в хорошем состоянии и ходили в детский сад, в то время как обычно в этом возрасте дети с этим заболеванием не могут даже говорить.
Различные варианты лечения в настоящее время изучаются в клинических испытаниях, в основном у пациентов позднего детского возраста. Эти методы лечения включают генную терапию, заместительную ферментную терапию (ФЗТ) и субстратную терапию (СЗТ).
Прогноз
Прогноз при метахроматической лейкодистрофии плохой. Наиболее частая форма заболевания, позднедетская, проявляется обычно на втором году жизни. Метахроматическая лейкодистрофия у таких детей обычно прогрессирует примерно в возрасте от 5 до 10 лет. Большинство детей с этой формой заболевания умирают в возрасте до 5 лет. Подростковая форма, начинающаяся в возрасте от четырех до подросткового возраста, протекает медленнее, чем поздняя младенческая форма, и симптомы могут появиться в возрасте от 10 до 20 лет; смерть обычно наступает через 10-20 лет после начала заболевания. Во взрослом возрасте первые симптомы обычно появляются в подростковом возрасте или позже. Для развития болезни может потребоваться 20–30 лет, хотя многие взрослые умирают в течение 6–14 лет после появления симптомов. В развитии этой формы могут быть периоды относительной стабильности и другие периоды более быстрого упадка.