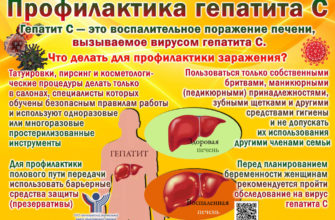
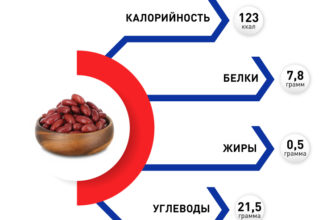
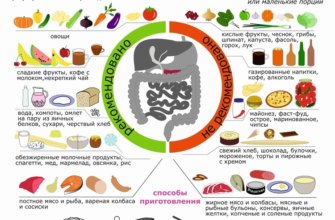
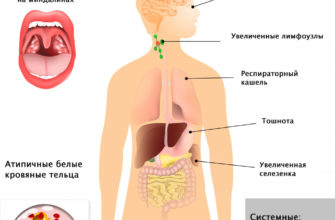
Что такое синдром Аперта?
Синдром Аперта (синдром Аперта или акроцефалосндактилия 1 типа) — редкое генетическое заболевание, возникающее при рождении. Больные с синдромом Аперта имеют характерные пороки развития черепа, лица, рук и ног.
Синдром Аперта характеризуется краниосиностозом, состоянием, при котором фиброзные соединения (швы) между костями черепа закрываются преждевременно. Это может повредить кости лица и сделать верхнюю часть головы заостренной. При заболевании могут соединяться пальцы рук и ног. Пострадавшие дети могут также иметь умственную отсталость. Тяжесть симптомов варьирует.
Синдром Аперта почти всегда является результатом новых генетических изменений (мутаций), возникающих случайно. Редко заболевание наследуется по аутосомно-доминантному типу. Пациенты с этим синдромом могут пройти симптоматическую терапию, включая реконструктивную хирургию черепа, лица, рук и ног.
Признаки и симптомы
Синдром Аперта характеризуется краниосиностозом, преждевременным закрытием фиброзных соединений (швов) между определенными костями черепа. У детей без краниосиностоза швы позволяют голове расти и увеличиваться. Затем кости сливаются, образуя череп.
У людей с краниосиностозом мозг продолжает расти после преждевременного закрытия швов.
Возникающее в результате давление по мере роста мозга может деформировать различные кости черепа и лица во время развития.
В зависимости от того, какие швы закрываются преждевременно, различается степень тяжести. У большинства пациентов происходит преждевременное закрытие швов между костями, образующими лоб и верхнюю часть черепа. Это приводит к тому, что голова становится заостренной с рождения (акроцефалия). Кроме того, задняя часть черепа может казаться уплощенной, с высоким и широким лбом (см. изображение выше). Череп может иметь большой «родничок» с поздним закрытием.
У некоторых пациентов также может развиться гидроцефалия, при которой спинномозговая жидкость скапливается в полостях головного мозга. Это может вызвать внутричерепное давление.
Краниосиностоз также может поражать кости лица. Это может привести к характерным дефектам лица. Пациенты с синдромом Аперта часто имеют широко расставленные глаза (гипертелоризм), выпученные глаза (экзофтальм) или косые глазные щели. Также у них может быть недоразвит центр лица (гипоплазия челюсти) и расщелина неба. Правая и левая стороны лица могут быть несимметричны.

У людей с этим заболеванием может быть уплощенный нос с низкой грудиной. У людей может быть задержка роста зубов, скученность зубов или открытый прикус.
Если отверстия между вашим носом и горлом сужены или заблокированы, или ваш хрящ трахеи поврежден, это может мешать дыханию и глотанию. Люди с этими проблемами могут иметь инфекции верхних дыхательных путей, апноэ во сне и недоедание.
Есть несколько характерных пороков развития рук и ног при синдроме Аперта. У пострадавших людей могут быть короткие, широкие и большие пальцы ног, которые выворачиваются наружу. У них также может быть частичное или полное соединение (синдактилия) некоторых пальцев рук и ног. У многих больных наблюдается полное сращение костей второго и четвертого пальцев и образование единого цельного ногтя (перчатковидная синдактилия). Однако возможны и другие комбинации.

Суставы пальцев ног становятся жесткими к четырем годам. В стопах синдактилия также поражает второй, третий и четвертый пальцы. Ногти на ногах могут быть частично сплошными или разделенными. Синдром обычно поражает верхние конечности в большей степени, чем нижние.
Синдром Аперта может поражать и другие системы органов (см. таблицу).
Причины
Синдром Аперта вызывается мутацией в гене рецептора 2 фактора роста фибробластов (FGFR2). Этот ген играет важную роль в развитии скелета. Гены предоставляют инструкции для создания белков, которые играют разные роли в организме. Когда ген мутирует, белковый продукт может работать неправильно. При синдроме Апера мутации в FGFR2 заставляют эти рецепторы неправильно связываться с факторами роста фибробластов. Это помогает мозгу формировать нормальные швы и может мешать многим другим структурам тела. Это аномальное образование является причиной пороков развития, наблюдаемых при синдроме Аперта.
Почти у всех описанных больных это заболевание было вызвано одной из двух специфических мутаций в гене FGFR2. (Эти мутации обозначены как «Ser252Trp» и «Pro253Arg».) Эти мутации могут вызывать немного другие симптомы, включая усиление синдактилии (сращивание пальцев). Различные мутации в гене FGFR2 могут вызывать ряд других связанных заболеваний, включая синдром Пфайффера, синдром Крузона и синдром Джексона-Вейсса.
У 95% пациентов синдром Апера вызван новой мутацией в гене FGFR2. Эти новые мутации появляются случайно по неизвестным причинам (спорадически). Сообщалось об отдельных случаях, связанных с увеличением возраста отца.
В редких случаях этот синдром наследуется по аутосомно-доминантному типу. Доминантные генетические нарушения возникают, когда для возникновения данного заболевания требуется только одна копия мутации. Риск передачи мутации от больного родителя потомству составляет 50% для каждой беременности. Риск одинаков для мужчин и женщин.
Затронутые группы населения
Подсчитано, что синдром Аперта поражает примерно одного из 65 000 новорожденных. Мужчины и женщины страдают в относительно равном количестве. С момента первоначального описания этого расстройства в 1894 и 1906 годах было зарегистрировано более 300 новых случаев. Больше всего случаев этого синдрома зарегистрировано у жителей Азии.
Диагностика
Диагноз чаще всего ставится при рождении или в младенчестве. Пациенты диагностируются посредством клинической оценки и различных специализированных тестов. Были выявлены физические симптомы, такие как лицевые аномалии или синдактилия.
Скелетные аномалии и врожденные пороки сердца выявляются с помощью методов визуализации, таких как компьютерная томография (КТ) или магнитно-резонансная томография (МРТ). Нарушение слуха можно обнаружить, исследуя слух вашего новорожденного.
Пациентов также можно проверить на наличие мутаций в гене FGFR2, что позволяет поставить генетический диагноз синдрома Апера.
В некоторых случаях симптомы синдрома Аперта можно обнаружить еще до рождения. Это можно сделать с помощью 2D или 3D пренатального УЗИ или магнитно-резонансной томографии (МРТ). Ультразвуковое исследование (УЗИ) – неинвазивная процедура, позволяющая получить изображение плода. Лечение позволяет выявить различия в форме черепа, аномалии строения лица и синдактилию. МРТ плода может предоставить больше информации о мозге плода, чем ультразвуковое сканирование.
Схожие по симптомам расстройства
Симптомы следующих расстройств могут быть сходными с симптомами синдрома Аперта. Сравнение может быть полезным в дифференциальной диагностике.
- Синдром Карпентера — редкое генетическое заболевание, связанное с краниосиностозом, полидактилией или синдактилией. Верхняя часть головы может казаться необычно заостренной (акроцефалия) или голова может казаться короткой и широкой (брахицефалия). Кроме того, черепные швы часто соединяются неравномерно, из-за чего голова и лицо кажутся асимметричными с одной стороны на другую. В некоторых случаях присутствуют дополнительные физические аномалии, такие как низкий рост, врожденный порок сердца, ожирение от легкой до умеренной степени, пупочная грыжа или крипторхизм. Многие люди с этим расстройством страдают от легкой до умеренной умственной отсталости.
- Синдром Крузона — редкое наследственное заболевание, связанное с краниосиностозом. Люди с синдромом Крузона также испытывают искажение середины лица, выпученные глаза и закупорку дыхательных путей, что вызывает затруднение дыхания и глотания. У некоторых пациентов очень большая голова (гидроцефалия). Синдром Крузона обычно не связан с умственной отсталостью или проблемами с руками, ногами, кистями или ступнями. Синдром Крузона вызывается изменениями в одном из генов FGFR, обычно FGFR2, и наследуется по аутосомно-доминантному типу.
- Синдром Джексона-Вейсса (СВД) — редкое наследственное заболевание, характеризующееся краниосиностозом и аномалиями нижних конечностей. Степень и тяжесть симптомов и признаков сильно различаются даже среди пораженных членов одной семьи. Первичные симптомы могут включать необычно плоскую, недоразвитую среднюю часть лица (гипоплазия средней части лица), аномально широкие большие пальцы ног и/или деформацию или сращение определенных костей стопы. МДД может быть спорадическим или наследоваться по аутосомно-доминантному типу.
- Синдром Пфайффера — редкое генетическое заболевание, характеризующееся краниосиностозом и аномально широкими и наклоненными медиальными большими и указательными пальцами. У большинства людей с этим заболеванием также большие глаза и кондуктивная тугоухость. Существует три типа синдрома Пфайффера. Типы II и III тяжелее. Синдром Пфайффера — аутосомно-доминантное заболевание, связанное с мутациями в генах FGFR2 и FGFR1.
Лечение
Лечение синдрома Аперта варьируется в зависимости от симптомов, которые есть у пациента. Для лечения может потребоваться помощь группы врачей-специалистов, в том числе педиатров и хирургов, нейрохирургов, врачей, специализирующихся на заболеваниях скелета, суставов и мышц (ортопедов), врачей уха, горла и носа (оториноларингологов) и врачей, специализирующихся на заболеваниях сердца. (кардиологи).
Специфические методы лечения синдрома Аперта носят симптоматический и поддерживающий характер. Краниосиностоз и гидроцефалия могут привести к аномально высокому давлению внутри черепа и головного мозга. В этих случаях может быть рекомендовано раннее оперативное вмешательство (через 2-4 мес после рождения) по коррекции краниосиностоза. У пациентов с гидроцефалией хирургическое вмешательство может также включать введение шунта для дренирования избытка спинномозговой жидкости (ЦСЖ) из головного мозга. Спинномозговая жидкость дренируется в другую часть тела, где она абсорбируется.
Для исправления черепно-лицевых дефектов могут быть рекомендованы корригирующие и реконструктивные операции. Хирургия также может помочь исправить полидактилию и синдактилию, а также другие дефекты скелета или физические отклонения. Пациентам с врожденными пороками сердца может потребоваться лечение некоторыми лекарствами, хирургическое вмешательство и/или другие терапевтические мероприятия. Некоторые люди с потерей слуха могут использовать слуховые аппараты.
Раннее вмешательство может иметь важное значение для обеспечения полного раскрытия потенциала детей с синдромом Аперта. Также помогут специальные терапевтические вмешательства, такие как физиотерапия, трудотерапия и специализированное обучение.
Генетическое консультирование рекомендуется больным людям и их семьям. Консультант-генетик сможет объяснить причины заболевания. Он или она может также обсудить возможность рождения новых детей с этим заболеванием. Кроме того, необходима психологическая поддержка для всей семьи.
Прогноз
Прогноз для детей с синдромом Апера зависит от того, насколько тяжела болезнь и какие системы организма поражены. Состояние может быть более серьезным, если оно влияет на дыхательную систему вашего ребенка или если внутри черепа накапливается давление, но эти проблемы можно исправить хирургическим путем.
У детей с этим синдромом часто возникают трудности с обучением. Некоторые дети страдают больше, чем другие.
Поскольку тяжесть заболевания может варьироваться в широких пределах, трудно предсказать ожидаемую продолжительность жизни. Заболевание может не оказывать существенного влияния на продолжительность жизни ребенка, особенно если у него нет пороков сердца.