Что такое амавроз Лебера?
Врожденный амавроз Лебера (известный как БАЛ) — редкое генетическое заболевание глаз. Пораженные дети часто слепы при рождении. Другие симптомы могут включать косоглазие; быстрые непроизвольные движения глаз (нистагм); необычная чувствительность к свету (светобоязнь); помутнение хрусталика (катаракта); и/или коническая форма переднего отрезка глаза (кератоз).
Врожденный амавроз Лебера начинается рано и может иметь самые тяжелые последствия. Уровень потери зрения при ВЭЛ варьирует от случая к случаю, но остается стабильным в 75% случаев. Около 15% детей имеют прогрессирующую потерю зрения, а у 10% может наблюдаться легкое, часто временное улучшение.
Амавроз Лебера характеризуется отсутствием каких-либо патологических изменений в глазу, которые можно распознать визуально, и заболевание обусловлено функциональным нарушением зрительного нерва наряду с другими отделами, отвечающими за зрение. Таким образом, в случае VAL глаза кажутся нормальными до первого осмотра.
Амавроз Лебера обычно наследуется как аутосомно-рецессивное генетическое заболевание.
Признаки и симптомы
Снижение зрительной реакции при рождении является первым симптомом заболевания. Часто ребенок тыкает, тыкает и трет глаза, чтобы стимулировать сетчатку к восприятию света. Эти действия могут привести к впадению или углублению глаз (энофтальм).
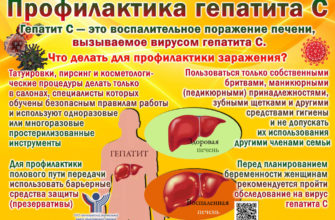
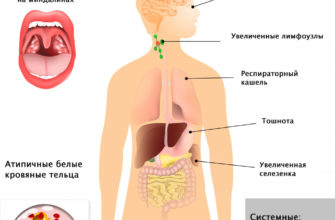
Другие симптомы могут включать:
Кроме того, у некоторых детей может быть потеря слуха, умственная отсталость и/или задержка развития.
Причины
Врожденный амавроз Лебера — моногенное заболевание, в которое вовлечено не менее 27 генов. Изменения (мутации) в этих генах могут составлять примерно 80-90% диагностированных случаев ВАЛ. Гены, ответственные за оставшиеся 10-20% диагнозов, неизвестны. ВАЛ обычно наследуется как аутосомно-рецессивное генетическое заболевание. Двадцать четыре VAL-ассоциированных гена вызывают заболевание только рецессивным образом. Известно, что два гена (IMPDH1 и OTX2) являются причиной доминирования болезни. Известно, что один ген (CRX) вызывает заболевание доминантным или рецессивным образом в зависимости от конкретной мутации.
Рецессивные генетические нарушения возникают, когда человек наследует две копии аномального гена одного признака, по одной от каждого родителя. Если у человека есть один нормальный ген и один ген болезни, этот человек будет носителем болезни, но обычно бессимптомно. Риск для двух родителей-носителей, которые оба передают дефектный ген и заражают ребенка, составляет 25% при каждой беременности. Риск рождения ребенка, который будет носителем, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит правильные гены от обоих родителей и будет генетически нормальным по этому конкретному признаку, составляет 25%. Риск одинаков для мужчин и женщин.
У человека около 20 000 различных генов, и у всех людей есть одна копия нескольких аномальных генов. Родители, являющиеся близкими родственниками (брат и сестра), с большей вероятностью имеют один и тот же аномальный ген, чем неродственные родители, что увеличивает риск рождения детей с рецессивным генетическим заболеванием.
В редких случаях амавроз Лебера наследуется как аутосомно-доминантное генетическое заболевание. В настоящее время известно, что мутации в трех генах, CRX, IMPDH1 и OTX2, связаны с этим типом VAL.
Доминантные генетические нарушения возникают, когда для возникновения данного заболевания требуется только одна копия аномального гена. Аномальный ген может быть унаследован от одного из родителей или может быть результатом новой мутации у больного человека. Риск передачи аномального гена от больного родителя потомству составляет 50% при любой беременности. Риск одинаков для мужчин и женщин.
Затронутые группы населения
Подсчитано, что заболеваемость ВАЛ составляет 1-2 случая на 100 000 рождений. Это расстройство поражает мужчин и женщин в равной степени.
Близкие по симптомам расстройства
Симптомы следующих нарушений могут быть сходны с симптомами врожденного амавроза Лебера. Сравнения могут быть полезны при дифференциальной диагностике:
- Синдром Сениора-Локена представляет собой редкое аутосомно-рецессивное генетическое заболевание, характеризующееся прогрессирующим истощением фильтрующего элемента почки (нефронофтизм) с медуллярной кистозной болезнью или без нее и прогрессирующей болезнью глаз. Это расстройство обычно появляется на первом году жизни.
- Синдром Жубера — это аутосомно-рецессивное наследственное генетическое заболевание, поражающее участки мозга, отвечающие за баланс и координацию. Это заболевание характеризуется специфическим МРТ-сканированием, называемым симптомом «корневого зуба», при котором червь мозжечка (пучки нервных волокон) отсутствует или недоразвит, а ствол головного мозга аномален. Наиболее распространенными симптомами синдрома Жубера являются отсутствие мышечного контроля (атаксия), аномальное дыхание (гиперпноэ), апноэ во сне, аномальные движения глаз и языка и низкий мышечный тонус.
- Нарушения биогенеза пероксисом в спектре синдрома Зеллвегера представляют собой группу редких, аутосомно-рецессивных, мультисистемных генетических нарушений, которые когда-то считались отдельными заболеваниями. Эти расстройства также известны как расстройства биогенеза пероксисом (PBD), группа расстройств, характеризующихся неспособностью организма производить пероксисомы. Синдром Цельвегера — самая тяжелая форма; неонатальная адренолейкодистрофия – промежуточная форма; а детская болезнь Рефсума — самая легкая форма. Нарушения биогенеза пероксисом в спектре синдрома Цельвегера могут поражать большинство органов тела. Неврологические расстройства, потеря мышечного тонуса (гипотония), потеря слуха, проблемы со зрением, дисфункция печени и аномалии почек являются общими симптомами этого расстройства.
Диагностика
Генетическое тестирование может быть проведено для определения наличия дефектного гена, вызывающего VAL. Это помогает оценить риск передачи заболевания от родителей к детям. Тесты также могут помочь вам поставить более точный диагноз, позволяя вам следить за новыми открытиями в интересующей вас области, исследовательскими разработками и новыми методами лечения.
Стандартные методы лечения
Лечение ВАЛ симптоматическое и поддерживающее. Генетическое консультирование рекомендуется семьям больных детей.
В 2017 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило генную терапию Luxturna (Люкстурна) для лечения детей и взрослых с двумя мутациями в гене RPE65. Luxturna производится компанией Spark Therapeutics, Inc.
Прогноз
Зрение обычно ухудшается с возрастом, а полная слепота наступает на третьем или четвертом десятилетии жизни.