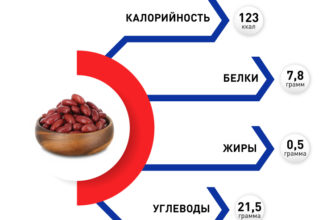
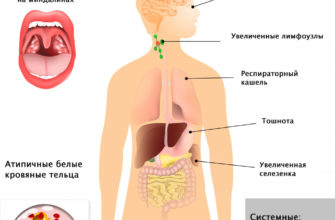
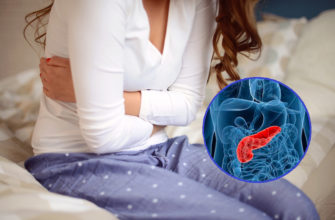
Что такое синдром Пейтца–Егерса?
Синдром Путц-Джегера (короче) является генетическим заболеванием, наследственным аутосомно-доминантным способом, происходящим примерно в 1/0000 до 1/200 000. Симптомы обычно появляются в первом десятилетии жизни и начинаются с веснушных пятен на темной коже (меланоцитарные пятна) вокруг рта, глаз, ноздрей, пальцев, внутри рта и вокруг заднего прохода. В возрасте около возраста пациентов многочисленные мягкие полипы, называемые Hamartoma, также начинают развиваться в пищеварительном тракте. Эти полипы расположены по всему пищеварительному тракту и могут вызвать тошноту, рвоту, боль в животе, обструкцию кишечника и ректальное кровотечение. Для удаления полипов (полипэктомия) могут потребоваться хирургия брюшной полости или эндоскопическое лечение для предотвращения связанных с полипов осложнений, таких как кишечные причины. Пациенты с синдромом Пьюц-Джегера имеют повышенный риск развития колоректального рака и других рака. Частые медицинские проверки и обследования необходимы для раннего обнаружения полипов и рака.
Синдром Путц-Джегера принадлежит к гетерогенной группе заболеваний, называемых синдромами из гамартоматических полиппитов, при которых растут многочисленные полипы в пищеварительном тракте пациентов. Впервые это было описано доктором Дж. Т. Коннора в 1895 году в паре сестер -близнецов с темными пигментными пятнами на губах и слизистой оболочкой рта. Эти симптомы были классифицированы как семейная команда в 1921 году, когда доктор Ян Пейц описал их четырех братьев и сестер. Эта команда была определена как отдельное расстройство доктором Гарольдом Эгерсом в 1949 году, когда он описал 10 случаев, а затем назвал синдром Пейца-Ферса в 1954 году доктором Андре Брюером. Генеральный ген (STK-11/LKB1) был идентифицирован в 1998 году и позволяет раннее обнаружение заболевания и проводясь скрининг у членов семьи.
Признаки и симптомы
Синдром Пейц-Джегера характеризуется возникновением многочисленных мягких полипов, называемых гамартомой, на слизистой оболочке пищеварительного тракта и темно-синими или темно-коричневыми пятнами с веснушками (меланоцитарные пятна) вокруг рта, глаза, ноздри, пальцы, слизистые Мембраны рта и заднего прохода. Эти меланоцитарные пятна могут появиться в первый год жизни и встречаются у большинства детей до пяти лет. Они склонны исчезать с возрастом и могут полностью исчезнуть во время полового созревания или в зрелом возрасте, хотя обычно они сохраняются в слизистой оболочке рта.
Полипы также начинают расти в первые годы жизни, но связанные с ними симптомы обычно появляются в возрасте от 10 до 30 лет. Около половины пациентов с этим расстройством должны подвергаться операции до 18 лет из -за сложностей, связанных с полипами. Полипы чаще всего развиваются в тонкой кишке (особенно в кишечнике), но они также могут возникать в желудке и толстой кишке. Редко полипы могут расти за пределами пищеварительного тракта и атакующих мочеточников, мочевого пузыря, легких, бронхов и желчного пузыря. Полипы желудочно -кишечного тракта могут вызвать боль в животе, рвоту, диарею, кишечную обструкцию и ректальное кровотечение, что может привести к анемии. Они также могут привести к потере кишечника вкусе (интуиция), что может привести к сильной боли в животе и необходимости выполнения операции в срочном режиме.
Люди с синдромом Пьюц-Северса находятся в группе с высоким риском рака желудочно-кишечного тракта и других рака, включая рак молочной железы, шейку матки, поджелудочную железу и легкие. Риск развития рака на протяжении всей их жизни у людей, пострадавших от этого заболевания, может составлять до 93%. Люди, у которых рак, обычно начинают заболеть около пятого десятилетия жизни (возраст 40-49 лет). У женщин, пострадавших от этого заболевания, существует повышенный риск получения легкой опухоли яичника, что симптомы могут быть нерегулярными или обильными менструацией или преждевременным сексуальным созреванием. Обычно, до 20 лет, у мужчин, затронутых этим заболеванием, может развиться опухоль в яичках, называемых клетками Celloli, которые выделяют эстроген и могут привести к увеличению молочной железы (Gynecomastia).
Причины
Синдром Пейца-Джегера является генетическим заболеванием, наследуемое аутосомно-доминантным способом, вызванным мутациями в гене STK11/LKB1. Доминирующие генетические расстройства возникают, когда необходима только одна копия аномального (патологического) гена. Аномальный ген может быть унаследован от одного из родителей или может быть результатом новой мутации от человека, пострадавшего от заболевания. Риск передачи аномального гена по болезни родителя составляет 50% во время каждой беременности. Риск такой же для мужчин и женщин.
Около 60-78% людей с этим расстройством имеют больных родственников. Около 80-94% пациентов с синдромом Пьюц-Северса существует мутация в гене STK 11, что означает, что другие гены также могут быть вовлечены в заболевание. Более 200 патогенных мутаций были зарегистрированы, и проникновение этих мутаций составляет 100%, что означает, что человек, который является носителем патогенной мутации, обречена на развитие заболевания.
Gen STK11 продуцирует белок, который участвует в регуляции деления клеток и запрограммированной гибели клеток (апоптоз). Это также влияет на p53, основной супрессор белок рака. Патогенные мутации в STK11 приводят к прекращению или нарушениям продукции белка геном и неконтролируемому росту клеток, что, в свою очередь, может привести к развитию мягких полипов (гамартома) и рака.
Считается, что пятна темного красителя (меланоцитарные пятна) вызваны воспалением и блокировкой миграции меланина из клеток, в которых он продуцируется (меланоциты), к клеткам, образующим наиболее внешний слой кожи (кератиноциты).
Затронутые группы населения
Синдром Путц-Эгерс-это редкое расстройство, которое в равной степени поражает мужчин и женщин и может происходить в любой расовой или этнической группе. Частота расстройств при рождении составляет от 1/0000 до 1/200 000. Ограниченные данные указывают на то, что это заболевание может происходить чаще в некоторых странах, таких как Нидерланды и Китай. Женщины в большей степени подвержены риску развития рака, чем мужчины, потому что синдром Пьюц-Джегера увеличивает вероятность развития молочной железы, яичника, шейки матки и матки.
Близкие по симптомам расстройства
Симптомы следующих заболеваний могут быть аналогичны симптомам пьюц-эгоистов. Сравнения могут быть полезны в дифференциальной диагностике:
- Молодежный полипный синдром является аутосомно -наследственным доминантным генетическим заболеванием, характеризующимся возникновением специфического типа гамартоматических полипов, называемых молодежными полипами. Большинство полипов легки, но люди, пострадавшие от этого заболевания, имеют повышенный риск развития колоректального рака и других рака.
- Синдром синагоги является аутосомно -наследственным доминантным генетическим заболеванием, характеризующимся появлением особого типа полипа, называемого обслуживаемым полипом (аденома), расположенного в большой кишке. Большинство полипов легки, но стоматологический полипонс связан с более частым появлением колоректального рака в личном и семейном интервью.
- Семейный агент Полипроксис (FAP) является редким синдромом наследственной предрасположенности к развитию рака, характеризующегося присутствием сотен или тысяч предраковых полипов (полипы аденомы). Если люди, пострадавшие от заболевания, не получают лечения, они неизбежно будут развивать колоректальный рак и/или прямую кишку в относительно молодом возрасте. APC наследуется аутосомно -доминантным способом и вызвана мутациями в гене APC.
- Синдром Турко является редким наследственным расстройством, характеризующимся возникновением легкого рака (полипов аденомы) слизистой оболочки желудочно -кишечного тракта и рака центральной нервной системы. Симптомы, связанные с образованием полипов, могут включать диарею, кровотечение из конечной части толстой кишки (прямая кишка), усталость, боль в животе и потерю веса. Пациенты также могут иметь неврологические симптомы, в зависимости от типа, размера и расположения опухоли головного мозга. Ученые считают, что синдром Тюрко представляет собой разнообразные семейные аденоколяты или синдром Линча (наследственное неполитичное колоректальное рак).
- Наследственная комбинация смешанной полиптичности представляет собой аутосомно -доминирующее генетическое заболевание, характеризующееся развитием нескольких типов полипов (атипичные молодежные полипы, гиперпластические полипы, глубокие аденомы и полипы аденомы) в пищеварительном тракте. Больники находятся в группе повышенного риска развития колоректального рака.
- Синдром опухоли PTEN-Gamartoma является спектром заболеваний, вызванных мутациями в гене-супрессоре опухоли PTEN. Эти расстройства характеризуются появлением многочисленных гамартомов, которые могут влиять на различные области тела. Симптомы также включают повышенный риск некоторых видов рака и расстройств нервной системы. Симптомы этого расстройства очень разные у разных людей и могут возникать в любом возрасте.
- Синдром кронхейта-канады (CCS) является чрезвычайно редким расстройством, характеризующимся появлением различных полипов кишечника, потерей вкуса, выпадения волос и проблем с ростом ногтей. SCC встречается в основном в популяции пожилых людей (средний возраст 59 лет) и обычно у мужчин. Считается, что SCC является болезнью, а не наследственным.
- Эндокринная неоплазия типа 2 (мужской тип 2) — это редкое генетическое заболевание, характеризующееся повышенным риском развития особой формы рака щитовидной железы (ядро рак щитовидной железы) и легких опухолей, охватывающих другие эндокринные железы. У людей с одним специальным типом синдрома, называемым мужчинами 2b, может развиться легкий рак, полученный из нервных клеток, называемых ганглионероматозом. Эти раковые заболевания возникают в пищеварительном тракте и могут вызвать вздутие живота, диарею, запор и неправильно увеличенные кишечники (мегаколон). Больники часто не набирают вес и не растут в ожидаемых темпах для их возраста и пола (гипоплазия).
- Синдром Карни — это редкое генетическое заболевание, характеризующееся возникновением многих мягких опухолей (множественных рака), чаще всего влияющих на сердце, кожа и эндокринная система, а также нарушения раскраски кожи (пигментация), что приводит к пятнам на коже в областях. Затронута болезнью. Легкий рак соединительной ткани (слизистые оболочки) распространены у людей с синдромом Карни и чаще всего возникают в сердце, где они могут вызвать серьезные, опасные для жизни осложнения, включая инсульт, венозные артерии или сердечную недостаточность. Синдром Карни может вызвать множество различных эндокринных расстройств разных желез. Другими раковыми заболеваниями являются опухоли кожи и нервных мембран (Schwvanoma). Расстройства пигментации кожи включают небольшие, плоские (веснушки) черные или коричневые пятна (множественные чечевицы) и маленькие, синие или синие пятна (синие знаки).
Диагностика
Клиническое распознавание синдрома Peutz-Seghers (PJS) может быть возведено, если какой-либо из следующих критериев соблюден:
- Присутствие как минимум двух полипов в команде Путц-Сигерс.
- Любое количество полипов и, по крайней мере, одного близкого родственника, у которого был диагностирован пютц-ферс.
- Характерные темные пигментные пятна (меланоцитарные пятна) и, по крайней мере, один близкий родственник, у которых был диагностирован SPE.
- Любое количество полипов и характерных темных пигментных пятен.
Меланоцитарные пятна могут быть обнаружены путем физического обследования. Полипы могут быть обнаружены с помощью эндоскопии, радиологического исследования или беспроводной капсулы эндоскопии, а при микроскопическом исследовании они классифицируются как полипы пьюц-Джегеров.
Генетические тесты для обнаружения патогенных мутаций в гене STK11 рекомендуются в случае любого из следующих критериев:
- Характерные темные пигментные пятна.
- Присутствие как минимум двух полипов SPE.
- Семейная внешность команды Peutz-Seghers.
Генетические тесты особенно полезны, когда патогенная мутация уже была идентифицирована в семье пациента. Считается, что проникновение мутации гена STK11 составляет 100% (что означает, что человек с патогенной мутацией имеет 100% вероятность заболевания), поэтому генетические тесты могут позволить тщательную диагностику до возникновения симптомов. Около половины пациентов с моча диагностируется благодаря генетическим тестам до возникновения симптомов.
Стандартные методы лечения
Пациенты должны сначала проконсультироваться с клиническим генетическим или генетическим консультантом. Поскольку нет никакого лекарства от синдрома Путц-Джегера, лечение в основном касается мониторинга и контроля симптомов. После первоначального диагноза у людей старше 8 лет или с симптомами рекомендуется провести эндоскопические тесты и тесты на тонкую кишку. Последнее может быть сделано с помощью магнитно-резонансной визуализации (магнитно-резонансное предприятие, МР-Ареография) или глотанием капсулы, которая производит внутренние желудочно-кишечные изображения (капсула видеодоскопия). Гинекологические тесты и тесты на грудь также рекомендуются для женщин старше 18 лет. Мужчинам рекомендуется проверить яички.
После предварительных скрининговых испытаний каждые 2-3 года эндоскопия, колоноскопия и исследование тонкой кишки для обнаружения полипов и потенциального рака. В случае женщин рекомендуются годовые маммографические тесты. Мужчины могут выполнять тестирование яичка яичка раз в два года.
Поскольку синдром Пютц-Северса увеличивает риск молочной железы, матки и яичников, женщины, затронутые этим заболеванием, могут подвергаться профилактической мастэктомии, гистерэктомии или сальфинго-ворттомии (хирургическое удаление молочной железы, матки, яичника и яичника).
Полипы, более 1 см, удаляются эндоскопически, чтобы избежать осложнений, связанных с полипами, такими как кровотечение и унизительное. Эти осложнения могут потребовать операции. Если пациент должен подвергнуться операции, эндоскопические полипы (полипэктомия) выполняются одновременно с операцией, чтобы снизить риск рецидива осложнений и хирургии.
В тех случаях, когда темные пятна красителя (меланоциты) оказывают очень негативное психологическое влияние на людей, затронутых заболеванием, их можно частично удалить с использованием лазерного лечения.
Прогноз
Peutz -Jeghers (PJS) не может быть полностью вылечено — это расстройство, которое длится целая жизнь, которая может быть унаследована. Люди с синдромом Путц-Джегера часто должны контролироваться для полипов. Эти полипы могут превратиться в рак или вызвать скопление, которое может потребовать операции.